
Exfoliation of zirconium aminophosphonates: investigation into their electronic structures by ab initio calculations†

Abstract
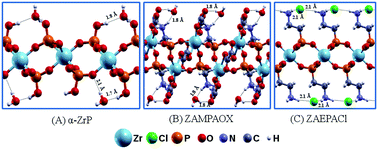
Recently, new structures of zirconium aminophosphonates were proposed by modifications of the synthesis of the known α-ZrP for different applications. In this work, we report a theoretical study of the structural and electronic properties of bulk materials and their 2D forms for three layered zirconium phosphonates: α-Zr(HPO4)2·H2O, Zr2H2[(C2O4)3(O3PCH2NH2)2]·2H2O (ZAMPAOX) and Zr(O3PCH2CH2NH3)2Cl2 (ZAEPACl). Moreover, the 13C and 31P NMR chemical shifts and infrared absorption spectra were also calculated and constructed to assist the characterization of future experimental analysis. For the 2D forms, the exfoliation energies were lower than 30 meV Å−2 and the energetic order was: α-ZrP and ZAMPAOX > ZAEPACl. This trend could be explained by the nature of the intra- and interlayer hydrogen bonds, which were analyzed by charge density difference plots. In terms of their band structures, all the materials presented indirect band gaps, though some differences related to band gap values were found after exfoliation. The influences of the hydration on their band structures were also evaluated. Structural modifications could be detected for the 2D forms of α-ZrP and ZAMPAOX after the removal of water molecules. The density of states suggested that the partial substitution of ions is a promising way to tune the band gap, as in the case of ZAEPACl with the substitution of chloride by other anions in the interlayer region.
- This article is part of the themed collection: Sustainability from intercalation compounds
 Please wait while we load your content...
Please wait while we load your content...

