
Unravelling the kinetics and molecular mechanism of the degenerate Cope rearrangement of bullvalene†

Abstract
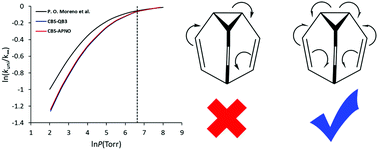
The kinetics and molecular mechanism of the gas phase degenerate Cope rearrangement (DCR) of bullvalene have been investigated by applying quantum mechanical calculations. Highly accurate energies (CBS-QB3 and CBS-APNO) and RRKM calculations were employed to study the kinetics and ‘fall-off’ behavior. It was found that the DCR of bullvalene (C3v) occurs through a bishomoaromatic transition structure (C2v) with an energy barrier of ∼49 kJ mol−1. The calculated activation energy and enthalpy were in good agreement with the available values in the literature, but lower than those of common Cope rearrangement; this result is related to the high stabilization energy due to the interaction of the allyl fragments in the bishomoaromatic transition structure. The fall-off curve revealed that TST breaks down slightly in estimating the high-pressure limit of the reaction rate and also obtaining the reaction as bimolecular is experimentally impossible. The synergic effect of ELF, NCI, and QTAIM has been used to study the molecular mechanism of the DCR of bullvalene at the B3LYP/6-311G(d,p) level of theory. The catastrophe sequence for DCR of bullvalene was 5-C†[F]2TS[F†]2C-0, which includes the following four steps: (i) homolytic rupture of the C1–C7 bond and formation of two pseudoradical centers on the C1 and C7 atoms; (ii) destruction of the pseudoradical centers; (iii) formation of new pseudoradical centers on the C3 and C5 atoms; and (iv) C-to-C coupling of the pseudoradical centers and formation of a new C3–C5 bond, where the electron density rearrangement takes place in an asynchronous manner.
 Please wait while we load your content...
Please wait while we load your content...

