
Adsorption characteristics and oxygen reduction reactions on pristine and Pt-, Co-decorated antimonenes: a DFT-D study
 a
Jianping
Sun,
*a
a
Jianping
Sun,
*a
Abstract
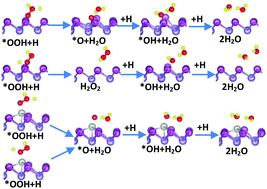
The adsorption characteristics of oxygen reduction reaction (ORR) intermediates on pristine and Pt-, Co-decorated antimonenes are studied using dispersion-corrected density functional theory (DFT-D). The most energy-favorable site of O atoms on pristine antimonene is the top site, with adsorption energy of 3.81 eV. OH, O2, and OOH tend to adsorb on the vacancy site, with adsorption energy of 2.43 eV, 1.59 eV and 1.04 eV, respectively. The most energy-favorable sites of Pt-, Co-decorated antimonenes are the valley sites, and the adsorption energies are 4.83 eV and 3.47 eV, respectively, which are higher than the binding energies to form dimers. After the antimonene is decorated with Pt, Co, all intermediates are adsorbed on the Pt, Co atom, with significant enhancement of adsorption strength. Furthermore, the ORR process is simulated, and the free energy changes and activation barriers in each step are calculated. There are both four-electron and two-electron paths on pristine antimonene, and the two-electron path is the dominant path. There are solely four-electron paths on Pt-, Co-decorated antimonenes. The free energy decline in each step on Pt-decorated antimonene is more even. However, from a kinetic perspective, there exists similarity in the OOR path on Pt-, Co-decorated antimonenes. The rate-determining steps are the reactions of the adsorbed O atoms reducing to OH on Pt-, Co-decorated antimonenes, and the activation barrier of the rate-determining step on Pt-decorated antimonenes is 0.27 eV lower than that on Co-decorated antimonenes. These findings provide a useful reference for the experimental exploration of monoatom-dispersed antimonenes as fuel cell cathodes.
 Please wait while we load your content...
Please wait while we load your content...

