
2D and 3D-QSAR, molecular docking and ADMET properties in silico studies of azaaurones as antimalarial agents
 *a
and
*a
and
Abstract
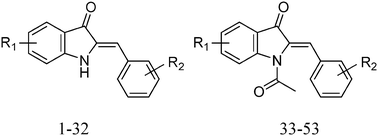
Malaria persists as the most infectious vector-borne disease in the world. The inhibition of the mitochondrial respiratory chain enzyme cytochrome bc1 has become the major focus as a molecular target in malarial parasites. Azaaurone derivatives have the potential to inhibit the mitochondrial respiratory chain of Plasmodium falciparum. Thus, with the aim of improving and proposing new molecules designed for antimalarial activity, we carried out a theoretical study of two-dimensional, three-dimensional-quantitative structure–activity relationships (QSAR) and docking analysis of a series of 53 azaaurones acting as inhibitors of the Qo site in cytochrome b. The generated 2D-QSAR model showed a good correlation coefficient of 0.871 and a very good prediction coefficient of the test set of 0.914. The predictive ability of the 2D-QSAR models was assessed using the Y-randomization test: internal (leave-one-out cross-validation) and external (test set with 11 compounds) validation. The 3D-QSAR model was generated using comparative molecular similarity indices analysis (CoMSIA). The best CoMSIA model with Q2 = 0.739, R2 = 0.844 and Rpred2 = 0.932 was evaluated using the Y-randomization test. The proposed models gave significant statistical quality. The results of molecular docking revealed that azaaurones specifically interact with the residues His183 and His82; this result was validated with a new validation criterion. Moreover, this is the first theoretical study showing the action of a molecule on the active site of heme bL. A good consistency between theoretical, 2D- and 3D-SQAR and molecular docking studies provided directions for the design of new drug candidates.
 Please wait while we load your content...
Please wait while we load your content...

