
Coligand driven diverse organometallation in benzothiazolyl-hydrazone derivatized pyrene: ortho vs. peri C–H activation†
 a
Sarat Chandra
Patra,
b
Subhadip
Roy,
c
Supriyo
Halder,
b
Thomas
Weyhermüller,
d
Kausikisankar
Pramanik
*b
and
Sanjib
Ganguly
*a
a
Sarat Chandra
Patra,
b
Subhadip
Roy,
c
Supriyo
Halder,
b
Thomas
Weyhermüller,
d
Kausikisankar
Pramanik
*b
and
Sanjib
Ganguly
*a
Abstract
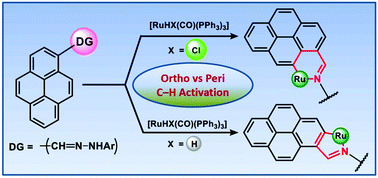
Benzothiazolyl hydrazones 1 (H2LPAH) incorporating polycyclic aromatic hydrocarbons (PAHs) have been fabricated as hemilabile scaffolds and elegantly utilized the inbuilt nitrogen donors as a proficient directing group (DG) to bring about ruthenium(II) assisted C–H activation in PAHs at both the peri and ortho positions. An isomeric pair of organometallics having formula [RuII(LPyr)(CO)(PPh3)2] (peri: 3a, ortho: 5a) have been conveniently prepared by varying the [RuII] precursors with H2LPyr. In contrast, only one type of activated product viz. [RuII(LAnc)(CO)(PPh3)2] 3b has been obtained with the 9-anthracene derivative of 1, H2LAnc, under analogous reaction conditions. The underlying mechanistic aspects have been elucidated by isolating the thermally unstable intermediates viz. [RuII(HLPyr)Cl(CO)(PPh3)2] 2a and [RuII(HLPyr)H(CO)(PPh3)2] 4a in the course of the peri and ortho C–H activation processes, respectively. The coligand (Cl/H) plays a vital role to bring about the C–H activation at the desired positions via formation of either a four- or five-membered metallacycle in 2a and 4a, respectively. The activation process vis-à-vis Ru–C bond formation in 3a can be achieved smoothly from 2a by a thermal transformation route, which proceeds via an initial rupture of the Ru–Nhydrazonyl bond. In contrast, the trans influential hydride coligand prefers a five-membered chelate to avoid confrontation with Nhydrazone in 4a, which in turn furnishes exclusively ortho activation owing to the close approach of the Ru–H bond towards ortho-H in pyrene. The organometallated complexes exhibit oxidative responses at mild potential. EPR and computational studies indicate that the redox activity originates from the ligand-centered orbitals. The observed rich optoelectronic features are analysed primarily as 1ILCT admixed with 1MLCT components by theoretical means, indicating an appreciable accumulation of electron density over the ligand backbone in their ground states.
 Please wait while we load your content...
Please wait while we load your content...

