
Synthesis, characterization, and anticancer activity of folate γ-ferrocenyl conjugates†

Abstract
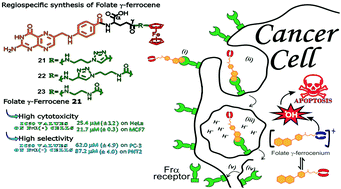
The quest for alternative therapeutic agents with safe and effective profiles is one of the biggest challenges in oncology. Folate-targeted therapy can deliver drugs selectively into malignant cells via the Frα receptor, whereas bioorganometallic chemistry offers potential new drug candidates. Herein, novel folate γ-ferrocene (γ-Fc) conjugates were synthesized through a regiospecific route by reacting amino-terminated glutamate (Glu) γ-Fc residues with pteroyl azide (68–78% yield), and their in vitro anticancer activities were evaluated on human cells. The Fc units were attached to protected Glu residues through different linkers using amide coupling and/or “click” reactions. Cyclic voltammetry and UV-vis measurements showed that the triazole group conjugated to the Fc donates electrons to the ferrous center, which facilitates the oxidation of Fc that is responsible for the antiproliferative activity. These results were confirmed by the higher activity of folate γ-triazoleFc 21, with IC50 values at 25.4 μM (±3.2) on HeLa and 21.7 μM (±0.3) on MCF-7 cells. This compound was up to 4-fold less toxic to healthy PNT2 cells and Frα-negative PC-3 cancer cells. Blocking experiments with an excess of free folic acid inhibited the folate activity suggesting their specific uptake via Frα. Glu residue analogs of folates, but lacking the pteroyl moiety, were much less toxic to cancer cells than folates. Hence, folate γ-Fcs are potential anticancer drug candidates due to their selectivity and enhanced cytotoxicity against Frα-positive malignant cells.
 Please wait while we load your content...
Please wait while we load your content...

