
Restoration of myocellular copper-trafficking proteins and mitochondrial copper enzymes repairs cardiac function in rats with diabetes-evoked heart failure†
 ab
Garth J. S.
Cooper
*abd
ab
Garth J. S.
Cooper
*abd
Abstract
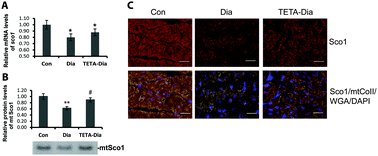
Diabetes impairs systemic copper regulation, and acts as a major independent risk factor for heart failure (HF) wherein mitochondrial dysfunction is a key pathogenic process. Here we asked whether diabetes might alter mitochondrial structure/function and thus impair cardiac performance by damaging myocellular pathways that mediate cell-copper homeostasis. We measured activity of major mitochondria-resident copper-enzymes cytochrome c oxidase (mt-Cco) and superoxide dismutase 1 (mt-Sod1); expression of three main mitochondrial copper-chaperones [Cco copper chaperone 17 (Cox17), Cox11, and mitochondria-resident copper chaperone for Sod1 (mt-Ccs)]; of copper-dependent Cco-assembly protein Sco1; and regulation of mitochondrial biogenesis, in left-ventricular (LV) tissue from groups of non-diabetic-control, untreated-diabetic, and divalent-copper-selective chelator-treated diabetic rats. Diabetes impaired LV pump function; ∼halved LV-copper levels; substantively decreased myocellular expression of copper chaperones, and enzymatic activity of mt-Cco and mt-Sod1. Divalent-copper chelation with triethylenetetramine improved cardiac pump function, restored levels of myocardial copper, the copper chaperones, and Sco1; and enzymatic activity of mt-Cco and mt-Sod1. Copper chelation also restored expression of the key mitochondrial biogenesis regulator, peroxisome-proliferator-activated receptor gamma co-activator-1α (Pgc-1α). This study shows for the first time that altered myocardial copper-trafficking is a key pathogenic process in diabetes-evoked HF. We also describe a novel therapeutic effect of divalent-copper-selective chelation, namely restoration of myocellular copper trafficking, which is thus revealed as a potentially tractable target for novel pharmacological intervention to improve cardiac function.
 Please wait while we load your content...
Please wait while we load your content...