
Choosing proper normalization is essential for discovery of sparse glycan biomarkers†
 *a
Lucija
Klarić,
bc
*a
Lucija
Klarić,
bc
Abstract
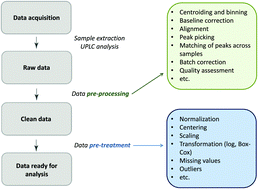
Rapid progress in high-throughput glycomics analysis enables the researchers to conduct large sample studies. Typically, the between-subject differences in total abundance of raw glycomics data are very large, and it is necessary to reduce the differences, making measurements comparable across samples. Essentially there are two ways to approach this issue: row-wise and column-wise normalization. In glycomics, the differences per subject are usually forced to be exactly zero, by scaling each sample having the sum of all glycan intensities equal to 100%. This total area (row-wise) normalization (TA) results in so-called compositional data, rendering many standard multivariate statistical methods inappropriate or inapplicable. Ignoring the compositional nature of the data, moreover, may lead to spurious results. Alternatively, a log-transformation to the raw data can be performed prior to column-wise normalization and implementing standard statistical tools. Until now, there is no clear consensus on the appropriate normalization method applied to glycomics data. Nor is systematic investigation of impact of TA on downstream analysis available to justify the choice of TA. Our motivation lies in efficient variable selection to identify glycan biomarkers with regard to accurate prediction as well as interpretability of the model chosen. Via extensive simulations we investigate how different normalization methods affect the performance of variable selection, and compare their performance. We also address the effect of various types of measurement error in glycans: additive, multiplicative and two-component error. We show that when sample-wise differences are not large row-wise normalization (like TA) can have deleterious effects on variable selection and prediction.
- This article is part of the themed collection: Glycomics & Glycoproteomics: From Analytics to Function
 Please wait while we load your content...
Please wait while we load your content...