
Machine-learning interatomic potentials enable first-principles multiscale modeling of lattice thermal conductivity in graphene/borophene heterostructures†
 *ab
*ab
Abstract
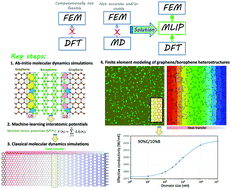
One of the ultimate goals of computational modeling in condensed matter is to be able to accurately compute materials properties with minimal empirical information. First-principles approaches such as density functional theory (DFT) provide the best possible accuracy on electronic properties but they are limited to systems up to a few hundreds, or at most thousands of atoms. On the other hand, classical molecular dynamics (CMD) simulations and the finite element method (FEM) are extensively employed to study larger and more realistic systems, but conversely depend on empirical information. Here, we show that machine-learning interatomic potentials (MLIPs) trained over short ab initio molecular dynamics trajectories enable first-principles multiscale modeling, in which DFT simulations can be hierarchically bridged to efficiently simulate macroscopic structures. As a case study, we analyze the lattice thermal conductivity of coplanar graphene/borophene heterostructures, recently synthesized experimentally (Sci. Adv., 2019, 5, eaax6444), for which no viable classical modeling alternative is presently available. Our MLIP-based approach can efficiently predict the lattice thermal conductivity of graphene and borophene pristine phases, the thermal conductance of complex graphene/borophene interfaces and subsequently enable the study of effective thermal transport along the heterostructures at continuum level. This work highlights that MLIPs can be effectively and conveniently employed to enable first-principles multiscale modeling via hierarchical employment of DFT/CMD/FEM simulations, thus expanding the capability for computational design of novel nanostructures.
- This article is part of the themed collections: Materials Horizons 10th anniversary regional spotlight collection: Europe and 2020 Materials Horizons most popular articles
 Please wait while we load your content...
Please wait while we load your content...

