
Stabilization of Cu+ by tuning a CuO–CeO2 interface for selective electrochemical CO2 reduction to ethylene†
 ‡c
Song
Hong,
a
Alex W.
Robertson,
d
Justus
Masa,
§e
Buxing
Han,
b
Yousung
Jung
*c
and
Zhenyu
Sun
*af
‡c
Song
Hong,
a
Alex W.
Robertson,
d
Justus
Masa,
§e
Buxing
Han,
b
Yousung
Jung
*c
and
Zhenyu
Sun
*af
Abstract
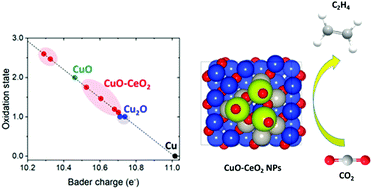
Electrochemical conversion of carbon dioxide (CO2) into multi-carbon fuels and chemical feedstocks is important but remains challenging. Here, we report the stabilization of Cu+ within a CuO–CeO2 interface for efficient and selective electrocatalytic CO2 reduction to ethylene under ambient conditions. Tuning the CuO/CeO2 interfacial interaction permits dramatic suppression of proton reduction and enhancement of CO2 reduction, with an ethylene faradaic efficiency (FE) as high as 50.0% at −1.1 V (vs. the reversible hydrogen electrode) in 0.1 M KHCO3, in stark contrast to 22.6% over pure CuO immobilized on carbon black (CB). The composite catalyst presents a 2.6-fold improvement in ethylene current compared to that of CuO/CB at similar overpotentials, which also exceeds many recently reported Cu-based materials. The FE of C2H4 remained at over 48.0% even after 9 h of continuous polarization. The Cu+ species are believed to be the adsorption as well as active sites for the activation of CO2 molecules, which remain almost unchanged after 1 h of electrolysis. Further density functional theory calculations demonstrate the preferred formation of Cu+ at the CuO–CeO2 interface. This work provides a simple avenue to convert CO2 into high-value hydrocarbons by rational stabilization of Cu+ species.
- This article is part of the themed collection: CO2 Utilisation
 Please wait while we load your content...
Please wait while we load your content...

