
Polymer principles behind solubilizing lignin with organic cosolvents for bioenergy†
 abc
Loukas
Petridis
*ab
abc
Loukas
Petridis
*ab
Abstract
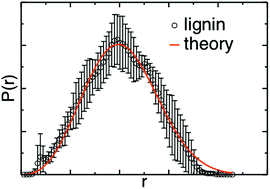
Lignin solubilization is key to a viable biorefinery because its removal leads to facile deconstruction of biomass and because the isolated lignin can serve to derive precursors of novel high-value materials. The mixing of organic solvents with water has been shown to improve biomass fractionation and lignin conversion reactions. However, generally-applicable solubilization strategies are lacking because of the remarkable variability of lignin across plant feedstocks. Here, to obtain a predictive understanding of lignin solvation, we perform molecular dynamics simulations of model lignin polymers in two mixtures of water with polar aprotic solvents: tetrahydrofuran (THF) : water and γ-valerolactone (GVL) : water. The model lignins include H-, G- and S-only homopolymers and lignins with an S : G 1 : 1 ratio. We find that a well-established theory of self-avoiding polymers in a “good” solvent describes accurately the physical conformations of all types of lignin in both solvents. As the degree of methoxy substitution increases in the homopolymers, the distributions of the lignin radius of gyration and the Flory exponent ν, which describes the lignin-solvent interactions, do not change in THF : water, while ν shifts to slightly higher values in GVL : water. We attribute this increase to the interaction between the methyl group of GVL with the lignin methoxy groups. We also find that the reduction in the lignin radius of gyration due to branching is accurately described by the Zimm–Stockmayer theory for both THF : water and GVL : water. The above findings validate the applicability of polymer physics concepts to lignin and suggest that GVL : water may have the most favorable interaction with S-lignin, whereas the interactions of THF : water with lignin are independent of lignin monomeric content.
 Please wait while we load your content...
Please wait while we load your content...

