
Variational calculations of excited states via direct optimization of the orbitals in DFT†

Abstract
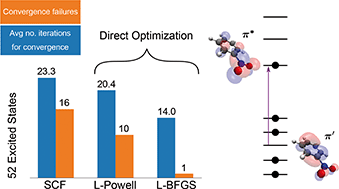
A direct optimization method for obtaining excited electronic states using density functionals is presented. It involves selective convergence on saddle points on the energy surface representing the variation of the energy as a function of the electronic degrees of freedom, thereby avoiding convergence to a minimum and corresponding variational collapse to the ground electronic state. The method is based on an exponential transformation of the molecular orbitals, making it possible to use efficient quasi-Newton optimization approaches. Direct convergence on a target nth-order saddle point is guided by an appropriate preconditioner for the optimization as well as the maximum overlap method. Results of benchmark calculations of 52 excited states of molecules indicate that the method is more robust than a standard self-consistent field (SCF) approach especially when degenerate or quasi-degenerate orbitals are involved. The method can overcome challenges arising from rearrangement of closely spaced orbitals in a charge-transfer excitation of the nitrobenzene molecule, a case where the SCF fails to converge. The formulation of the method is general and can be applied to non-unitary invariant functionals, such as self-interaction corrected functionals.
- This article is part of the themed collection: New horizons in density functional theory
 Please wait while we load your content...
Please wait while we load your content...

