
London dispersion forces without density distortion: a path to first principles inclusion in density functional theory†
Derk Pieter
Kooi  *a
and
Paola
Gori-Giorgi
a
*a
and
Paola
Gori-Giorgi
a
*a
and
Paola
Gori-Giorgi
a
Abstract
We analyse a path to construct density functionals for the dispersion interaction energy from an expression in terms of the ground state densities and exchange–correlation holes of the isolated fragments. The expression is based on a constrained search formalism for a supramolecular wavefunction that is forced to leave the diagonal of the many-body density matrix of each fragment unchanged, and is exact for the interaction between one-electron densities. We discuss several aspects: the necessary features of a density functional approximation for the exchange–correlation holes of the monomers, the optimal choice of the one-electron basis (named “dispersals”), and the functional derivative with respect to monomer density variations.
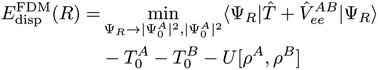
- This article is part of the themed collection: New horizons in density functional theory
 Please wait while we load your content...
Please wait while we load your content...

