
A multiscale modelling approach to elucidate the mechanism of the oxygen evolution reaction at the hematite–water interface†

Abstract
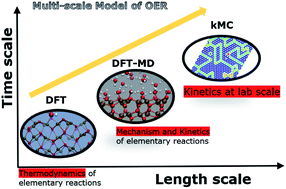
Photoelectrochemical (PEC) splitting of water to make hydrogen is a promising clean-energy technology. The oxygen evolution reaction (OER) largely determines the energy efficiency in PEC water-splitting. Hematite, which is a cheap and sustainable semiconductor material with excellent chemical properties, a favourable band gap (2.1 eV) and composed of earth abundant elements is a suitable model photoanode material for studying OER. To understand the design of energy efficient anodes, it is highly desirable to have mechanistic insight into OER at an atomistic level which can be directly connected to experimentally measured quantities. We present a multiscale computational model of OER which connects the thermodynamics and kinetics of elementary charge transfer reactions in OER to kinetics of OER at laboratory length and time scales. We couple density functional theory (DFT) and DFT based molecular dynamics (DFT-MD) simulations with solvent effects at an atomistic level with kinetic Monte Carlo (kMC) simulations at a coarse-grained level in our multiscale model. The time and applied bias potential dependent surface coverage, which are experimentally not known, and the O2 evolution rate during OER at the hematite–water interface are calculated by the multiscale model. Furthermore, the multiscale model demonstrates the effect of explicitly modelling the interaction of water with the electrode surface via direct adsorption.
- This article is part of the themed collection: Reaction mechanisms in catalysis
 Please wait while we load your content...
Please wait while we load your content...

