
Interpreting Tafel behavior of consecutive electrochemical reactions through combined thermodynamic and steady state microkinetic approaches†

Abstract
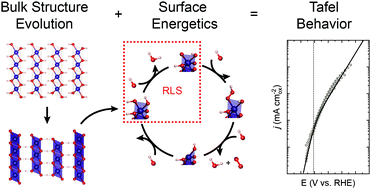
Assessing the reaction pathway of multi-electron-transfer reactions is an essential yet difficult task for the rational design of electrocatalysts. In this work, we develop a heuristic approach that combines thermodynamic adsorption energetics calculated through density functional theory with microkinetic modeling using the steady state approximation to interpret the potential-dependent Tafel behavior of consecutive electrochemical reactions. In doing so, we introduce a kinetic framework for ab initio calculations that ensures self-consistent adsorption energetics based on kinetically limited adsorbate coverages. The approach is applied to experimental results on CoOx(OH)2−x single crystal electrocatalyst particles yielding coverage dependent mechanistic information and identification of the rate-limiting step with standard rate constants for the oxygen evolution reaction on the (11![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 0) surfaces of the β-Co(OH)2, β-CoOOH, and CoO2 bulk phases. This generalizable method enables catalyst benchmarking based on determining the active species involved and associated intrinsic reaction rate constants in consecutive multi-electron-transfer reactions.
0) surfaces of the β-Co(OH)2, β-CoOOH, and CoO2 bulk phases. This generalizable method enables catalyst benchmarking based on determining the active species involved and associated intrinsic reaction rate constants in consecutive multi-electron-transfer reactions.
 Please wait while we load your content...
Please wait while we load your content...

