
A theoretical mechanistic study of IrIII/CuI-metallaphotoredox catalyzed asymmetric radical decarboxylative cyanation†

Abstract
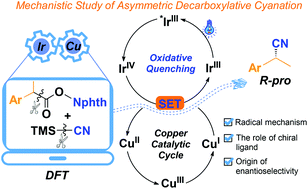
Visible-light-induced asymmetric metallaphotoredox catalysis has become a powerful strategy in synthetic organic chemistry. IrIII/CuI dual asymmetric catalysis has been developed to achieve enantioselective decarboxylative cyanation. However, detailed mechanisms, such as catalytic cycles for dual catalysts and the role of a chiral ligand, remain obscure in these reactions. In this study, the catalytic cycle of this reaction is systematically investigated by DFT calculations to clarify the quenching mechanism of the photocatalyst and the origin of the excellent enantioselectivity. Interestingly, the radical mechanism merging oxidative quenching (IrIII–*IrIII–IrIV–IrIII) and copper catalytic cycles (CuI–CuII–CuIII–CuI) is favourable. It consists of five major processes: single-electron oxidation of *IrIII by N-hydroxy-phthalimide (NHP) esters followed by decarboxylation to generate benzyl radical, oxidation of CuI by IrIVvia a single-electron transfer (SET) process, cyanide exchange, radical capture by CuII, and C–CN reductive elimination from CuIII. The cyanide exchange is the rate-determining step, whereas the C–CN reductive elimination is the enantio-determining step of the reaction. In addition, the origin of the high enantioselectivity was analyzed from the steric and electronic effects. This study will hopefully benefit the future understanding of such photoredox-mediated dual catalyzed asymmetric synthesis.
- This article is part of the themed collection: Spotlight Collection: Photoinduced redox chemistry
 Please wait while we load your content...
Please wait while we load your content...

