
Probing the potential of metalla-N-heterocyclic carbenes towards activation of enthalpically strong bonds†

Abstract
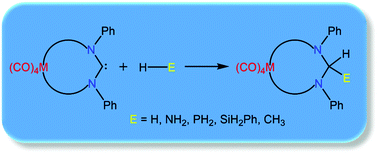
Density functional theory calculations are employed to explore the reactivity of metalla-N-heterocyclic carbenes (MNHCs) towards activation of a variety of small molecules (H2, NH3, PH3, SiH3Ph and CH4). All the MNHCs considered are found to have a stable singlet ground state and possess suitable electronic properties for their application in small molecule activation. The calculated energy barriers of E–H (E = H, C, N, Si, P) activation for the MNHCs are found to be in agreement with those of the experimentally evaluated cyclic alkyl(amino)carbene (CAAC) and diamidocarbenes (DACs), thereby indicating the activating effect of the incorporation of an ancillary metal center within a cyclic NHC, and highlighting a new, underexplored strategy in achieving difficult bond activations with carbenes.
 Please wait while we load your content...
Please wait while we load your content...

