
Determining the coordination environment and electronic structure of polymer-encapsulated cobalt phthalocyanine under electrocatalytic CO2 reduction conditions using in situ X-Ray absorption spectroscopy†
 ‡a
Aniruddha
Deb,
‡ab
Kwan Yee
Leung,
a
Weixuan
Nie,
a
William S.
Dean,
a
James E.
Penner-Hahn
*ab
and
Charles C. L.
McCrory
*ac
‡a
Aniruddha
Deb,
‡ab
Kwan Yee
Leung,
a
Weixuan
Nie,
a
William S.
Dean,
a
James E.
Penner-Hahn
*ab
and
Charles C. L.
McCrory
*ac
Abstract
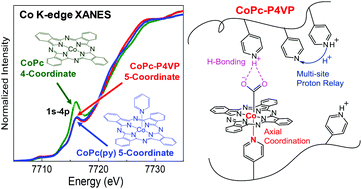
Encapsulating cobalt phthalocyanine (CoPc) within the coordinating polymer poly-4-vinylpyridine (P4VP) results in a catalyst–polymer composite (CoPc–P4VP) that selectively reduces CO2 to CO at fast rates at low overpotential. In previous studies, we postulated that the enhanced selectively for CO over H2 production within CoPc–P4VP compared to the parent CoPc complex is due to a combination of primary, secondary, and outer-coordination sphere effects imbued by the encapsulating polymer. In this work, we perform in situ electrochemical X-ray absorption spectroscopy measurements to study the oxidation state and coordination environment of Co as a function of applied potential for CoPc, CoPc–P4VP, and CoPc with an axially-coordinated py, CoPc(py). Using in situ X-ray absorption near edge structure (XANES) we provide experimental support for our previous hypothesis that Co changes from a 4-coordinate square-planar geometry in CoPc to a mostly 5-coordinate species in CoPc(py) and CoPc–P4VP. The coordination environment of CoPc–P4VP is potential-independent but pH-dependent, suggesting that the axial coordination of pyridyl groups in P4VP to CoPc is modulated by the protonation of the polymer. Finally, we show that at low potential the oxidation state of Co in the 4-coordinate CoPc is different from that in the 5-coordinate CoPc(py), suggesting that the primary coordination sphere modulates the site of reduction (metal-centered vs. ligand centered) under catalytically-relevant conditions.
- This article is part of the themed collection: New Talent: Americas
 Please wait while we load your content...
Please wait while we load your content...

