
Accurate computed spin-state energetics for Co(iii) complexes: implications for modelling homogeneous catalysis†

Abstract
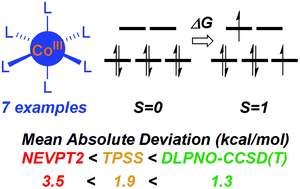
Co(III) complexes are increasingly prevalent in homogeneous catalysis. Catalytic cycles involve multiple intermediates, many of which will feature unsaturated metal centres. This raises the possibility of multi-state character along reaction pathways and so requires an accurate approach to calculating spin-state energetics. Here we report an assessment of the performance of DLPNO-CCSD(T) (domain-based local pair natural orbital approximation to coupled cluster theory) against experimental 1Co to 3Co spin splitting energies for a series of pseudo-octahedral Co(III) complexes. The alternative NEVPT2 (strongly-contracted n-electron valence perturbation theory) and a range of density functionals are also assessed. DLPNO-CCSD(T) is identified as a highly promising method, with mean absolute deviations (MADs) as small as 1.3 kcal mol−1 when Kohn–Sham reference orbitals are used. DLPNO-CCSD(T) out-performs NEVPT2 for which a MAD of 3.5 kcal mol−1 can be achieved when a (10,12) active space is employed. Of the nine DFT methods investigated TPSS is the leading functional, with a MAD of 1.9 kcal mol−1. Our results show how DLPNO-CCSD(T) can provide accurate spin state energetics for Co(III) species in particular and first row transition metal systems in general. DLPNO-CCSD(T) is therefore a promising method for applications in the burgeoning field of homogeneous catalysis based on Co(III) species.
 Please wait while we load your content...
Please wait while we load your content...

