
Impacts of hydrogen bonding interactions with Np(v/vi)O2Cl4 complexes: vibrational spectroscopy, redox behavior, and computational analysis†

Abstract
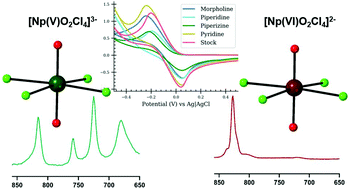
The neptunyl (Np(V)O2+/Np(VI)O22+) cation is the dominant form of 237Np in acidic aqueous solutions and the stability of the Np(V) and Np(VI) species is driven by the specific chemical constituents present in the system. Hydrogen bonding with the oxo group may impact the stability of these species, but there is limited understanding of how these intermolecular interactions influence the behavior of both solution and solid-state species. In the current study, we systematically evaluate the interactions between the neptunyl tetrachloride species and hydrogen donors in coordination complexes and in the related aqueous solutions. Both Np(V) compounds (N2C4H12)2[Np(V)O2Cl4]Cl (Np(V)pipz) and (NOC4H10)3[Np(V)O2Cl4] (Np(V)morph) exhibit directional hydrogen bonding to the neptunyl oxo group while Np(VI) compounds (NC5H6)2[Np(VI)O2Cl4] (Np(VI)pyr) and (NOC4H10)4[Np(VI)O2Cl4]·2Cl (Np(VI)morph) assemble via halogen interactions. The Raman spectra of the solid-state phases indicate the activation of vibrational bands when there is asymmetry of the neptunyl bond, while these spectral features are not observed within the related solution phase spectra. Density functional theory calculations of the Np(V)pipz system suggest that activation of the ν3 asymmetric stretch and other combination modes lead to additional complexity within the solid-state spectra. Electrochemical analyses of complexes in the solution phases are consistent with the results of the crystallization experiments as the voltammetric potentials of Np(V)/Np(VI) complexes in the presence of protonated heterocycles differ from the potentials of pure Np(V) and may correlate with the hydrogen bonding interactions.
 Please wait while we load your content...
Please wait while we load your content...

