
Substituent-dependent generation of tricyclic frameworks by the rhodium-catalyzed cycloisomerization of homopropargyl allene-alkynes: a theoretical study†

Abstract
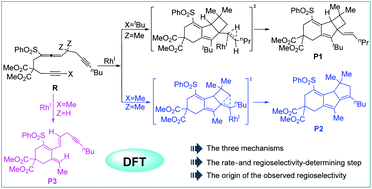
Polycyclic compounds having biological activities can be modified by employing different substituents. Substituent-dependent generation of tricyclic frameworks by the rhodium-catalyzed cycloisomerization of homopropargyl allene-alkynes was investigated by using density functional theory calculations. A mechanistic study revealed that all the three reactions, A (Z = Me, X = tBu), B (Z = Me, X = Me), and C (Z = H, X = Me), underwent initial oxidative cyclization, from which the divergent cycloisomerization was triggered when different substituents were employed. It was found from our calculations that reactions A and B underwent alkyne insertion into the Rh–C(sp2) bond and resulted in a key metal carbenoid intermediate. From the metal carbenoid species, reaction A bearing a more hindered tert-butyl group prefers 1,2-hydrogen migration to produce a 6/5/4 tricyclic product, while reaction B bearing a less hindered methyl group prefers 1,2-carbon migration leading to ring expansion and producing a 6/5/5 tricyclic product. The origins of the distinct selectivity were disclosed, which are beneficial for the development of novel related reactions. For reaction C (Z=H, Z=H or Z=H, Z=Me), the β-hydrogen elimination was found to be favored over the alkyne insertion into the Rh–C(sp2) bond and thus the monocyclic product is delivered. The E/Z effects involved in the above two cases were also discussed.
 Please wait while we load your content...
Please wait while we load your content...

