
A subtle structural change in the distal haem pocket has a remarkable effect on tuning hydrogen peroxide reactivity in dye decolourising peroxidases from Streptomyces lividans†
 a
Florian S. N.
Dworkowski,
b
Dimitri A.
Svistunenko,
a
Michael A.
Hough
a
and
Jonathan A. R.
Worrall
*a
a
Florian S. N.
Dworkowski,
b
Dimitri A.
Svistunenko,
a
Michael A.
Hough
a
and
Jonathan A. R.
Worrall
*a
Abstract
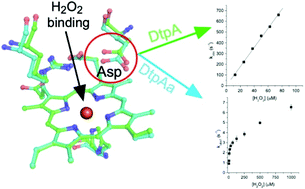
Dye decolourising peroxidases (DyPs) are oxidative haem containing enzymes that can oxidise organic substrates by first reacting with hydrogen peroxide. Herein, we have focused on two DyP homologs, DtpAa and DtpA, from the soil-dwelling bacterium Streptomyces lividans. By using X-ray crystallography, stopped-flow kinetics, deuterium kinetic isotope studies and EPR spectroscopy, we show that both DyPs react with peroxide to form compound I (a FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species and a porphyrin π-cation radical), via a common mechanism, but the reactivity and rate limits that define the mechanism are markedly different between the two homologs (DtpA forms compound I rapidly, no kinetic isotope effect; DtpAa 100-fold slower compound I formation and a distinct kinetic isotope effect). By determining the validated ferric X-ray structure of DtpAa and comparing it with the ferric DtpA structure, we attribute the kinetic differences to a subtle structural repositioning of the distal haem pocket Asp side chain. Through site-directed mutagenesis we show the acid–base catalyst responsible for proton-transfer to form compound I comprises a combination of a water molecule and the distal Asp. Compound I formation in the wild-type enzymes as well as their distal Asp variants is pH dependent, sharing a common ionisation equilibrium with an apparent pKa of ∼4.5–5.0. We attribute this pKa to the deprotonation/protonation of the haem bound H2O2. Our studies therefore reveal a mechanism for compound I formation in which the rate limit may be shifted from peroxide binding to proton-transfer controlled by the distal Asp position and the associated hydrogen-bonded water molecules.
O species and a porphyrin π-cation radical), via a common mechanism, but the reactivity and rate limits that define the mechanism are markedly different between the two homologs (DtpA forms compound I rapidly, no kinetic isotope effect; DtpAa 100-fold slower compound I formation and a distinct kinetic isotope effect). By determining the validated ferric X-ray structure of DtpAa and comparing it with the ferric DtpA structure, we attribute the kinetic differences to a subtle structural repositioning of the distal haem pocket Asp side chain. Through site-directed mutagenesis we show the acid–base catalyst responsible for proton-transfer to form compound I comprises a combination of a water molecule and the distal Asp. Compound I formation in the wild-type enzymes as well as their distal Asp variants is pH dependent, sharing a common ionisation equilibrium with an apparent pKa of ∼4.5–5.0. We attribute this pKa to the deprotonation/protonation of the haem bound H2O2. Our studies therefore reveal a mechanism for compound I formation in which the rate limit may be shifted from peroxide binding to proton-transfer controlled by the distal Asp position and the associated hydrogen-bonded water molecules.
- This article is part of the themed collection: Inorganic Reaction Mechanisms
 Please wait while we load your content...
Please wait while we load your content...

