
A DFT and microkinetic study of propylene oxide selectivity over copper-based catalysts: effects of copper valence states†

Abstract
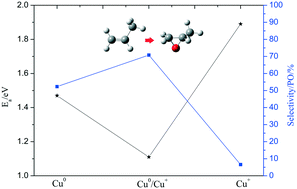
The development of high-performance copper-based catalysts is critical for the selective oxidation of propylene in both technology and scientific fields. To ably understand the effect of valence states between Cu and Cu2O on the selectivity and activity of propylene oxide (PO), density functional theory (DFT) calculations were carried out to investigate the reaction mechanisms over Cu(111), Cu2O(111) and Cu0/Cu+ interface catalysts, corresponding to Cu0, Cu+ and Cu0/Cu+ active sites. Herein, two reaction routes, namely, dehydrogenation (AHS) and epoxidation pathways (OMMP1 and OMMP2), are taken into account in the catalytic mechanism for propylene oxidation with atomic oxygen as the oxidant. According to the microkinetic model analysis, the Cu2O(111) surface is least active and has only 6.56% selectivity for PO with a higher apparent activation energy of 1.89 eV. On the Cu(111) surface, it was found that the Cu0 site is favorable for propylene epoxidation, thus leading to a moderate selectivity for PO (52.3%), and the apparent activation energy is 1.47 eV. A comparison with the corresponding bulk experimental data shows good agreement with the modeling data for Cu(111) and Cu2O(111). Compared with other two catalysts, the Cu0/Cu+ interface exhibits highest selectivity and activity for PO, and the selectivity for PO can reach up to 70.8% and at the same time the apparent activation energy is only 1.11 eV, which is lower than Cu(111) and Cu2O(111) surfaces. The present work indicated that the Cu0/Cu+ site is the most favorable for propylene epoxidation and the PO formation selectivity and activity follow the trend of Cu0/Cu+ > Cu0 > Cu+. The relationship of selectivity and activity with the copper valence effect could supply a clue for designing Cu-based catalysts with high efficiency for propylene epoxidation.
 Please wait while we load your content...
Please wait while we load your content...

