
Influence of spin state and electron configuration on the active site and mechanism for catalytic hydrogenation on metal cation catalysts supported on NU-1000: insights from experiments and microkinetic modeling†
 a
Omar K.
Farha,
*e
Massimiliano
Delferro,
*c
Alex B. F.
Martinson
*d
and
Rachel B.
Getman
*a
a
Omar K.
Farha,
*e
Massimiliano
Delferro,
*c
Alex B. F.
Martinson
*d
and
Rachel B.
Getman
*a
Abstract
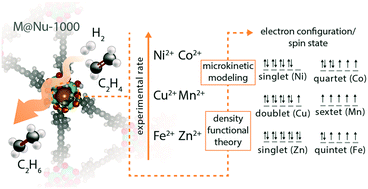
The mechanism of ethene hydrogenation to ethane on six dicationic 3d transition metal catalysts is investigated. Specifically, a combination of density functional theory (DFT), microkinetic modeling, and high throughput reactor experiments is used to interrogate the active sites and mechanisms for Mn@NU-1000, Fe@NU-1000, Co@NU-1000, Ni@NU-1000, Cu@NU-1000, and Zn@NU-1000 catalysts, where NU-1000 is a metal–organic framework (MOF) capable of supporting metal cation catalysts. The combination of experiments and simulations suggests that the reaction mechanism is influenced by the electron configuration and spin state of the metal cations as well as the amount of hydrogen that is adsorbed. Specifically, Ni@NU-1000, Cu@NU-1000, and Zn@NU-1000, which have more electrons in their d shells and operate in lower spin states, utilize a metal hydride active site and follow a mechanism where the metal cation binds with one or more species at all steps, whereas Mn@NU-1000, Fe@NU-1000, and Co@NU-1000, which have fewer electrons in their d shells and operate in higher spin states, utilize a bare metal cation active site and follow a mechanism where the number of species that bind to the metal cation is minimized. Instead of binding with the metal cation, catalytic species bind with oxo ligands from the NU-1000 support, as this enables more facile H2 adsorption. The results reveal opportunities for tuning activity and selectivity for hydrogenation on metal cation catalysts by tuning the properties that influence hydrogen content and spin, including the metal cations themselves, the ligands, the binding environments and supports, and/or the gas phase partial pressures.
 Please wait while we load your content...
Please wait while we load your content...

