
Theoretical studies on the N–X (X = Cl, O) bond activation mechanism in catalytic C–H amination†

Abstract
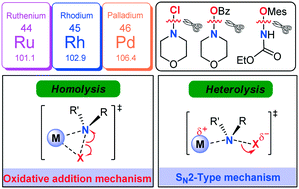
A comprehensive DFT study has been performed on the mechanism of Rh-catalysed C–H amination of N-arylbenzamide with N-chloromorpholine. The catalytic cycle starting from the N–H deprotonation of N-arylbenzamide was ruled out due to the inconsistency between the calculated and experimentally observed rate-determining steps. Instead, the CsOAc induced pre-deprotonated form of N-arylbenzamide participates in the catalytic cycle. The reaction then goes through C–H activation via concerted metalation–deprotonation, N–Cl cleavage, reductive elimination, and protonation to complete the catalytic cycle. It has been found that an SN2-type N–Cl cleavage fashion is more favorable compared with the conventional oxidative addition manner. Both cleavage manners of the N–Cl bond were manifested by geometry structure and orbital population analyses. More importantly, such an SN2-type mechanism also works for Ru-catalysed N–Cl cleavage and Rh-/Ru-/Pd-catalysed N–O cleavage involved in C–H amination systems. It is found that the less geometrical distortion accounts for the preference towards the SN2-type. It is theoretically demonstrated that the manner of N–O cleavage is affected by the ligand substituents. Modification of N-benzoyloxymorpholine with an electron-withdrawing substituent could facilitate the SN2-type N–O cleavage process. Such an SN2-type N–X (X = N, O) cleavage process was established for the first time in the field of C–H amination, which enriches the C–H amination chemistry.
 Please wait while we load your content...
Please wait while we load your content...

