
First-principles studies on α-Fe2O3 surface slabs and mechanistic elucidation of a g-C3N4/α-Fe2O3 heterojunction†

Abstract
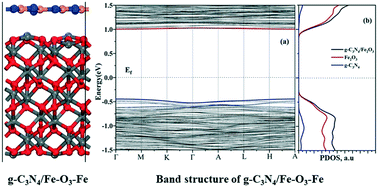
Amongst metal oxides, hematite (α-Fe2O3) is a promising photocatalytic anode material, and thus improving the mechanistic understanding of its surface and electronic properties is an important task. Here, first-principles density-functional theory calculations were carried out to explore the surface energies and electronic properties of α-Fe2O3 surface slabs and polar Fe2O3 surfaces, examining several competing terminations of the α-Fe2O3 (0001) surface modeled via slabs with increasing surface area from 12 Fe (21.983 Å2) to 48 Fe-atom (87.92 Å2) slabs. Although there were quantitative differences observed in surface energies from 12 Fe to 48 Fe layers, the lowest-energy α-Fe2O3 (0001) surface was observed at all oxygen chemical potentials. The influence of Fe- or O-termination on the fundamental semiconducting or metallic nature was analysed; among the surface slabs, we considered that four have non-zero band gaps (semiconductors), and two are metallic. Further, to enhance the photocatalytic activity of Fe2O3, an alternative effective strategy was to examine Fe2O3-based heterostructures. These were modelled, and to model the heterojunctions/heterostructures, the lowest-energy and most stable AFM surface slabs were chosen to act as heterojunctions in contact with another semiconductor g-C3N4. For the mono-layer of the g-C3N4 and Fe–O3–Fe surface heterojunction, DFT+U calculations were performed to explore the density of states (DOS), charge-density differences, local potentials and the band offset. These studies reveal that the g-C3N4/Fe–O3–Fe heterojunction is type I – a straddling-gap type rather than a staggered type. Further, we investigated the g-C3N4 convergence by placing two and three layers of g-C3N4 on the Fe–O3–Fe surface.
 Please wait while we load your content...
Please wait while we load your content...

