
The origin of different driving forces between O–H/N–H functional groups in metal ligand cooperation: mechanistic insight into Mn(i) catalysed transfer hydrogenation†

Abstract
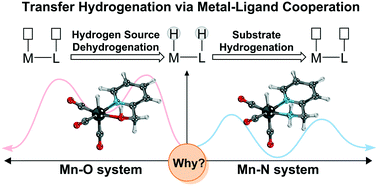
Metal–ligand cooperation catalysis is currently the prevailing strategy in the field of homogeneous catalyst research, and is widely used in direct catalytic hydrogenation and transfer hydrogenation reactions. Herein, a density functional theory (DFT) study is conducted to clarify the origin of the different activities of Mn(I) bifunctional catalysts bearing similar Lewis base functional ligands, and amine and hydroxyl groups. The results indicate that a Mn(I) catalyst with an OH group as a bifunctional group requires a higher activation free energy barrier relative to the catalyst with amine as an active ligand, which is in line with the experimental observations. By comparing the electronic structures of the key intermediates in the two catalytic systems, it is found that the Mn–O complex catalyst is thermodynamically unstable and may lead to irreversible decomposition, which accounts for its lower catalytic activity. Moreover, the inductive effect between the OH group and the metal hydride increases unfavorable orbital interactions in the Mn–O system. Consequently, the generation of a metal hydride intermediate becomes a thermodynamically uphill process, further leading to a lack in driving force for the dehydrogenation of iPrOH. Further investigation suggests that the driving force of the catalyst can be tuned by changing the different oxidation states of the metal centers, revealing a crucial role for the metal center in M–L bond cooperation mode in MLC catalysis. This study highlights that although the hydroxyl and amine groups are both Lewis base functional ligands, subtle differences in the electronic effects of ligand have a significant impact on the activities of the metal–ligand cooperation (MLC) catalysts.
 Please wait while we load your content...
Please wait while we load your content...

