
Mononuclear Fe in N-doped carbon: computational elucidation of active sites for electrochemical oxygen reduction and oxygen evolution reactions†

Abstract
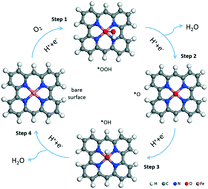
Non-precious metal catalysts are well investigated in electrocatalysis. Fe and N co-doped carbon (Fe–N–C) catalysts have drawn great attention due to their low cost and good performance in the oxygen reduction reaction (ORR) and recently in the oxygen evolution reaction (OER). Based on the recent advances of a variety of physical characterization techniques, more information about the chemical environment of the catalytic active sites has been acquired. However, due to the complexity of the catalytic material and process, the real active structures are still controversial. In this work, several active sites are proposed for Fe–N–C catalysts (L-FeNx, x = 2 or 4, L = nothing, O or OH) and their performance in electrochemical ORR and the OER are investigated. The computations are based on density functional theory (DFT), including Van der Waals interaction and solvation effect with an implicit electrolyte model. Calculations indicate that the catalytic activity of the Fe centers depends strongly on the N coordination number and on the presence of extra ligands like OH group. In particular, HO–FeN2, but not FeN4, appears as the most active site. Scaling relations are obtained by connecting the free energy of potential-determining steps with the adsorption free energy of intermediates. Furthermore, three promising active sites suggested from scaling relations are studied by the more elaborate surface charging approach, which includes the influence of the applied potential and the electrolyte. The results show that the specific treatment of the influence of the applied potential has a minor influence at low potential, which is the case for the ORR, but a major influence at higher potential, as for the OER, changing the calculated overpotential by up to 0.34 V.
- This article is part of the themed collection: 2020 Catalysis Science & Technology Hot Articles
 Please wait while we load your content...
Please wait while we load your content...

