
Role of hemibonding in the structure and ultraviolet spectroscopy of the aqueous hydroxyl radical†
 a
and
John M.
Herbert
*a
a
and
John M.
Herbert
*a
Abstract
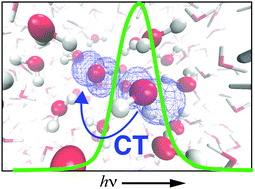
The presence of a hemibond in the local solvation structure of the aqueous hydroxyl radical has long been debated, as its appearance in ab initio simulations based on density functional theory is sensitive to self-interaction error (favoring a two-center, three-electron hemibond) but also to finite-size effects. Simulations reported here use a mixed quantum mechanics/molecular mechanics (QM/MM) framework in a very large periodic simulation cell, in order to avoid finite-size artifacts and to facilitate testing of various density functionals, in order to probe the effects of delocalization error. The preponderance of hemibonded structures predicted by generalized gradient approximations persists in simulations using the hybrid functionals B3LYP and PBE0, but is reduced to a minor population if the fraction of exact exchange is increased to 50%. The hemibonded population is also small in simulations employing the long-range corrected functional LRC-ωPBE. Electronic spectra are computed using time-dependent density functional theory, and from these calculations emerges a consensus picture in which hemibonded configurations play an outsized role in the absorption spectrum, even when present as a minority species. An intense 1b2(H2O) → 2pπ(˙OH) charge-transfer transition in hemibonded configurations of the radical proves to be responsible for an absorption feature at 230 nm that is strongly shifted with respect to the gas-phase absorption at 307 nm, but this intense feature is substantially diminished in aqueous geometries where the hemibond is absent. Although not yet sufficient to quantitatively establish the population of hemibonded ˙OH(aq), these simulations do suggest that its presence is revealed by the strongly shifted ultraviolet absorption spectrum of the aqueous radical.
 Please wait while we load your content...
Please wait while we load your content...

