
Effects of structural variations on π-dimer formation: long-distance multicenter bonding of cation-radicals of tetrathiafulvalene analogues†

Abstract
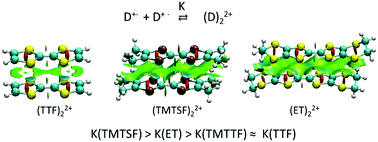
The multicenter (pancake) bonding between cation-radicals of tetramethyltetraselenafulvalene, TMTSF+˙, tetramethyltetrathiafulvalene, TMTTF+˙, and bis(ethylenedithio)-tetrathiafulvalene, ET+,˙ was compared to that of tetrathiafulvalene, TTF+˙. To minimize counter-ion effects, the cation-radical salts with weakly coordinating anions (WCA), tetrakis(3,5-trifluoromethylphenyl)borate, dodecamethylcarborane and hexabromocarborane were prepared. Solid-state (X-ray and EPR) measurements revealed diamagnetic π-dimers in the TMTSF and ET salts and the separate monomers in the TTF salts with all WCAs, while TMTTF existed as a dimer in one and a monomer in two salts. The variable-temperature UV-Vis studies of these salts in solution showed that the thermodynamics of formation of the π-bonded dimers of TMTTF+˙ was close to that of TTF+˙, while TMTSF+˙ and ET+˙ showed a higher propensity for π-dimerization. These data indicated that the replacement of sulfur with heavier selenium or insertion of ethylenedithia-substituents into the TTF core increases the π-dimers’ stability. Yet, computational analysis indicated that the weakly covalent component of π-bonding decreases in the order TTF > TMTTF > TMTSF > ET. The higher stability of the π-dimers of TMTSF+˙ and ET+˙ cation-radicals was related to a decrease of the electrostatic repulsion between cationic counter-parts and an increase of dispersion components in these associations.
 Please wait while we load your content...
Please wait while we load your content...

