
Electronic and spin structures of CaMn4Ox clusters in the S0 state of the oxygen evolving complex of photosystem II. Domain-based local pair natural orbital (DLPNO) coupled-cluster (CC) calculations using optimized geometries and natural orbitals (UNO) by hybrid density functional theory (HDFT) calculations†

Abstract
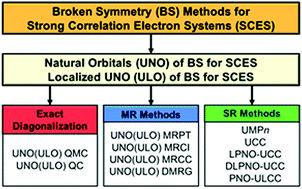
Domain-based local pair natural orbital (DLPNO) coupled cluster single and double (CCSD) with triple perturbation (T) correction methods were performed to elucidate the relative stabilities of ten different intermediate structures of the CaMn4Ox cluster in the S0 state of the oxygen evolving complex (OEC) of photosystem II (PSII). Full geometry optimizations of all the S0 intermediates were performed by the UB3LYP-D3/Def2-TZVP methods, providing the assumed geometrical structures and starting natural orbitals (UNO) for DLPNO-CCSD(T)/Def2TZVP calculations. The effective exchange integrals (J) for the spin Hamiltonian models for the ten intermediates were obtained by the UB3LYP/Def2-TZVP calculations followed by the general spin projections. DLPNO-CCSD(T) calculations followed by the CBS extrapolation procedure elucidated that the (II, III, IV, IV) and (III, III, III, IV) valence states in the CaMn4O5 cluster of the OEC of the PS II were nearly degenerated in energy in the S0 state, indicating an important role of dynamical electron correlation effects for the valence and spin fluctuations in strongly correlated electron systems (SCESs) consisting of 3d transition metals.
- This article is part of the themed collection: Quantum Theory: The Challenge of Transition Metal Complexes
 Please wait while we load your content...
Please wait while we load your content...

