
Structure, stability, infrared spectra, and bonding of OHm(H2O)7 (m = 0, ±1) clusters: ab initio study combining the particle swarm optimization algorithm†
 a
Nina
Ge
c
a
Nina
Ge
c
Abstract
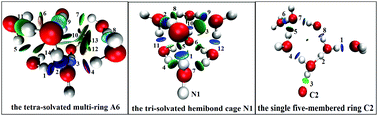
The various structural candidates of anionic, neutral, and cationic water clusters OHm(H2O)7 (m = 0, ±1) have been globally predicted by combining the particle swarm optimization method and quantum chemical calculations. Geometry optimization and vibrational analysis for the optimal structures were performed with the MP2/aug-cc-pVDZ method, and the energy profile was further refined at the CCSD(T)/CBS level. Special attention was paid to the relationships between configurations and energies, particularly the first solvation shell coordination number of OH− and OH. For OH−(H2O)7, OH(H2O)7, and OH+(H2O)7 clusters, the most stable species at room temperature are predicted to be the tetra-solvated multi-ring structure A6, the tri-solvated hemibond cage structure N1, and the single five-membered ring structure C2, respectively. The temperature effects on the stability of these three systems were also explored via Gibbs free energies. Furthermore, for the OH−(H2O)7 clusters, the assignments of vibrational transitions in the OH stretching region are in good agreement with the studies of small hydroxide ion-water clusters, and the IR spectra of two isomers (tetra-solvated multi-ring A6 and penta-solvated cage A3) may match future experimental observation well. By topological analysis and reduced density gradient analysis, the structural characteristics and bonding strengths of the studied clusters were investigated. This work indicates the excellent performance of the PSO search algorithm and CALYPSO on water clusters, and may further provide extensive insights into the chemical behavior such as the transport mechanism of OH− ions and OH radicals in the aqueous phase.
 Please wait while we load your content...
Please wait while we load your content...

