
Disclosing the microscopic mechanism and adsorption properties of CO2 capture in N-isopropylethylenediamine appended M2(dobpdc) series†
 *a
Eric
Ganz
c
*a
Eric
Ganz
c
Abstract
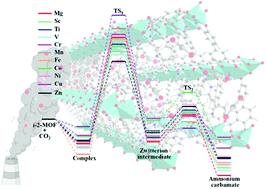
The detailed picture of the microscopic mechanism for CO2 capture in N-isopropylethylenediamine (i-2) functionalized M2(dobpdc) (dobpdc4− = 4,4′-dioxidobiphenyl-3,3′-dicarboxylate; M = Mg, Sc–Zn) has been determined for the first time via systematic computations with van der Waals (vdW) corrected density functional theory (DFT) methods. The results show that acting as a Lewis base, the i-2 molecule can strongly interact with the acidic open metal sites of M2(dobpdc) via its primary amine with binding energies of 132 to 178 kJ mol−1 for different metals. After exposure to gaseous CO2, CO2 is captured by inserting into the metal–N bond. The corresponding CO2 binding energies (43–69 kJ mol−1) vary depending on the metal centers. i-2-Sc2(dobpdc) and i-2-Mg2(dobpdc) with high CO2 binding energies have promising potential for CO2 capture. Moreover, the results demonstrate that the CO2 capture process involves two steps, consisting of simultaneous nucleophilic attack of the CO2 onto the metal-bound N atom with proton transfer. This results in the formation of a zwitterion intermediate (step1), and then rearrangement of the zwitterion intermediate into the final product ammonium carbamate (step2). The first step with relatively high barriers (0.99–1.49 eV) is rate-determining. The second step with low barriers (less than 0.50 eV) can easily occur and will promote the reaction. This work uncovers the complicated microscopic mechanism of CO2 capture with i-2 functionalized MOFs at the molecular level. This study provides fundamental understanding of the adsorption process and insights into the design and synthesis of highly efficient CO2 capture materials.
 Please wait while we load your content...
Please wait while we load your content...

