
Assessing cluster models of solvation for the description of vibrational circular dichroism spectra: synergy between static and dynamic approaches†
 a
Jessica
Bowles,
b
Sascha
Jähnigen,
c
Carine
Clavaguéra,
b
Florent
Calvo,
d
Rodolphe
Vuilleumier
c
and
Anne
Zehnacker
*a
a
Jessica
Bowles,
b
Sascha
Jähnigen,
c
Carine
Clavaguéra,
b
Florent
Calvo,
d
Rodolphe
Vuilleumier
c
and
Anne
Zehnacker
*a
Abstract
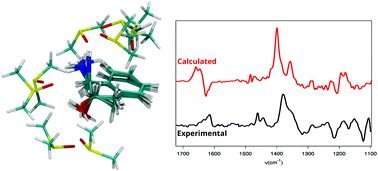
Solvation effects are essential for defining the shape of vibrational circular dichroism (VCD) spectra. Several approaches have been proposed to include them into computational models for calculating VCD signals, in particular those resting on the “cluster-in-a-liquid” model. Here we examine the capabilities of this ansatz on the example of flexible (1S,2S)-trans-1-amino-2-indanol solvated in dimethyl sulfoxide (DMSO). We compare cluster sets obtained from static calculations with results from explicit molecular dynamics (MD) trajectories based on either force field (FF) or first-principles (FP) methods. While the FFMD approach provides a broader sampling of configurational space, FPMD and time-correlation functions of dipole moments account for anharmonicity and entropy effects in the VCD calculation. They provide a means to evaluate the immediate effect of the solvent on the spectrum. This survey singles out several challenges associated with the use of clusters to describe solvation effects in systems showing shallow potential energy surfaces and non-covalent interactions. Static structures of clusters involving a limited number of solvent molecules satisfactorily capture the main effects of solvation in the bulk limit on the VCD spectra, if these structures are correctly weighted. The importance of taking into consideration their fluxionality, i.e. different solvent conformations sharing a same hydrogen bond pattern, and the limitations of small clusters for describing the solvent dynamics are discussed.
- This article is part of the themed collections: PCCP Perspectives and 2020 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

