
A computational study of Tat–CDK9–Cyclin binding dynamics and its implication in transcription-dependent HIV latency†
 *a
*a
Abstract
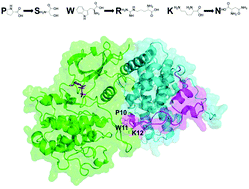
HIV is a virus that attacks the T cells. HIV may either actively replicate or become latent within host cells for years. Since HIV uses its own protein Tat to hijack the host CDK9–Cyclin complex for transcription, Tat is implicated in transcription-dependent HIV latency. To quantify the impact of Tat binding, we propose a computational framework to probe the dynamics of the CDK9–Cyclin interface and the ATP pocket reorganization upon binding by different Tat mutants. Specifically, we focus on mutations at three Tat residues P10, W11, and N12 that are known to interact directly with CDK9 based on the crystal structure of the Tat–CDK9–Cyclin complex. Our molecular dynamics simulations show that the CDK9–Cyclin interface becomes slightly weaker for P10S and W11R mutants but tighter for the K12N mutant. Furthermore, the side chain orientation of residue K48 in the ATP pocket of CDK9 is similar to the inactive state in P10S and W11R simulations, but similar to the active state in K12N simulations. These are consistent with some existing but puzzling observations of latency for these mutants. This framework may hence help gain a better understanding of the role of Tat in the transcription-dependent HIV latency establishment.
 Please wait while we load your content...
Please wait while we load your content...

