
Mechanistic insight into H2-mediated Ni surface diffusion and deposition to form branched Ni nanocrystals: a theoretical study†

Abstract
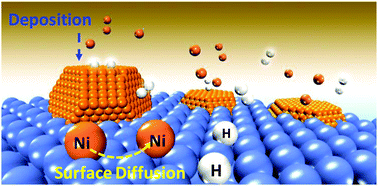
Present work systematically investigates the kinetic role played by H2 molecules during Ni surface diffusion and deposition to generate branched Ni nanostructures by employing Density Functional Theory (DFT) calculations and ab initio molecule dynamic (AIMD) simulations, respectively. The Ni surface diffusion results unravel that in comparison to the scenarios of Ni(110) and Ni(100), both the subsurface and surface H hinder the Ni surface diffusion over Ni(111) especially under the surface H coverage of 1.5 ML displaying the lowest Ds values, which greatly favors the trapping of the adatom Ni and subsequent overgrowth along the 〈111〉 direction. The Ni deposition simulations by AIMD further suggest that both the H2 molecule (in solution) and surface dissociatively adsorbed atomic H can promote Ni depositions onto Ni(111) and Ni(110) facets in a liquid solution. Moreover, a cooperation effect between H2 molecules and surface atomic H can be clearly observed, which greatly favors Ni depositions. Additionally, in addition to working as the solvent, the liquid C2H5OH can also interact with the Ni(111) surface to produce the surface atomic H, which then favored the Ni deposition. Finally, the Ni deposition rate predicted using the deposition constant (Ddep) was found to be much higher than its surface diffusion rate predicted using Ds for Ni(111) and Ni(110), which quantitatively verified the overgrowth along the 〈111〉 and 〈110〉 directions to produce the branched Ni nanostructures.
 Please wait while we load your content...
Please wait while we load your content...

