
Molecular dynamics simulation of the formation of Cu–Pt nanocontacts in the mechanically controlled break junction experiments†
 *ab
*ab
Abstract
The formation of the Cu–Pt nanocontacts has been investigated by means of classical molecular dynamics simulations. The simulations of the mechanically controlled break junction experiment have been performed in wide ranges of temperatures (0–300 K) and at relative Pt concentrations (0–20%). The structure of the breaking area has been studied 2 ns before the final breaking of the nanocontacts. The length of the breaking area increases with the increase of the temperature and decreases with the increase of the relative Pt concentration. The structure of the breaking area has been investigated by means of the radial distribution function method. The breaking area usually has one of the following structures: (i) a bulk-like structure, (ii) a structure consisting of centered icosahedrons rotated 90°, or (iii) an icosahedral structure composed of pentagonal rings. The structure of the breaking area is almost independent of the temperature and the stretching direction due to the strong Cu–Pt interaction.
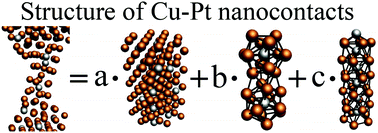
 Please wait while we load your content...
Please wait while we load your content...

