
Accurate elemental boiling points from first principles†
 *ab
and
*ab
and
Abstract
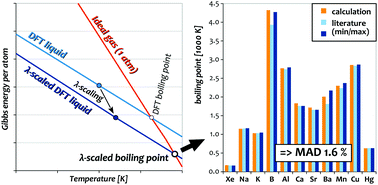
The normal boiling point (NBP) is a fundamental property of liquids and marks the intersection of the Gibbs energies of the liquid and the gas-phase at ambient pressure. This work provides the first comprehensive demonstration of the calculation of boiling points of atomic liquids through first-principles molecular-dynamics simulations. To this end, thermodynamic integration (TDI) and perturbation theory (TPT) are combined with a density-functional theory (DFT) Hamiltonian, which provides absolute Gibbs energies, internal energies, and entropies of atomic liquids with an accuracy of a few meV/atom. Linear extrapolation to the intersection with the Gibbs energy of a non-interacting gas-phase eventually pins-down the NBPs. While these direct results can already be quite accurate, they are susceptible to a systematic over or underbinding of the employed density functional. It is shown how this dependency can be strongly reduced and the robustness of the method increased through a simple linear correction termed λ-scaling. Eventually, by carefully tuning of the technical parameters of the approach, the walltime per element is reduced from weeks to about a day (10–20k core-hours), enabling extensive testing for B, Al, Na, K, Ca, Sr, Ba, Mn, Cu, Xe, and Hg. This comprehensive benchmark demonstrates the excellent performance and robustness of the approach with a mean absolute deviation (MAD) of less than 2% from experimental NBPs and very similar accuracy for liquid entropies (MAD 2.3 J (mol K)−1, 2% relative). In some cases, the uncertainties in the predictions are several times smaller than the variation between literature values, allowing us to clear out long-standing ambiguities in the NBPs of B and Ba.
 Please wait while we load your content...
Please wait while we load your content...

