
Macroscopic quantum electrodynamics and density functional theory approaches to dispersion interactions between fullerenes†

Abstract
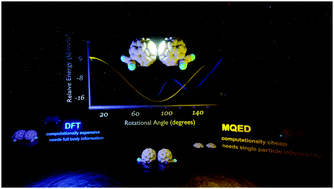
The processing and material properties of commercial organic semiconductors, for e.g. fullerenes is largely controlled by their precise arrangements, specially intermolecular symmetries, distances and orientations, more specifically, molecular polarisabilities. These supramolecular parameters heavily influence their electronic structure, thereby determining molecular photophysics and therefore dictating their usability as n-type semiconductors. In this article we evaluate van der Waals potentials of a fullerene dimer model system using two approaches: (a) Density Functional Theory and, (b) Macroscopic Quantum Electrodynamics, which is particularly suited for describing long-range van der Waals interactions. Essentially, we determine and explain the model symmetry, distance and rotational dependencies on binding energies and spectral changes. The resultant spectral tuning is compared using both methods showing correspondence within the constraints placed by the different model assumptions. We envision that the application of macroscopic methods and structure/property relationships laid forward in this article will find use in fundamental supramolecular electronics.
 Please wait while we load your content...
Please wait while we load your content...

