
An ab initio investigation of alkali–metal non-covalent bonds B⋯LiR and B⋯NaR (R = F, H or CH3) formed with simple Lewis bases B: the relative inductive effects of F, H and CH3†

Abstract
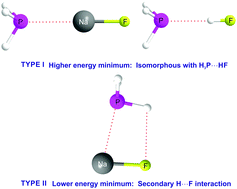
The alkali–metal bonds formed by simple molecules LiR and NaR (R = F, H or CH3) with each of the six Lewis bases B = OC, HCN, H2O, H3N, H2S and H3P were investigated by ab initio calculations at the CCSD(T)/AVTZ and CCSD(T)/awCVTZ levels of theory with the aim of characterising this type of non-covalent interaction. In some complexes, two minima were discovered, especially for those involving the NaR. The higher-energy minimum (referred to as Type I) for a given B was found to have geometry that is isomorphous with that of the corresponding hydrogen-bonded analogue B⋯HF. The lower-energy minimum (when two were present) showed evidence of a significant secondary interaction of R with the main electrophilic region of B (Type II complexes). Energies DCBSe for dissociation of the complexes into separate components were found to be directly proportional to the intermolecular stretching force constant kσ The value of DCBSe could be partitioned into a nucleophilicity of B and an electrophilicity of LiR or NaR, with the order ELiH ≳ ELiF = ELiCH3 for the LiR and ENaF > ENaH ≈ ENaCH3 for the NaR. For a given B, the order of the electrophilicities is ELiR > ENaR, which presumably reflects the fact that Li+ is smaller than Na+ and can approach the Lewis base more closely. A SAPT analysis revealed that the complexes B⋯LiR and B⋯NaR have larger electrostatic contributions to De than do the hydrogen- and halogen-bonded counterparts B⋯HCl and B⋯ClF.
 Please wait while we load your content...
Please wait while we load your content...

