
Examining the degradation of environmentally-daunting per- and poly-fluoroalkyl substances from a fundamental chemical perspective†
 a
and
Gabriel L. C.
de Souza
*ab
a
and
Gabriel L. C.
de Souza
*ab
Abstract
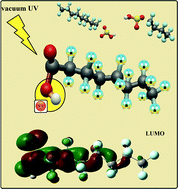
In this work, ground and excited-state properties were used as descriptors for probing mechanisms as well as to assess potential alternatives for tackling the elimination of perfluorobutane sulfonic acid (PFBS) – C4F9SO2OH, perfluorooctane sulfonic acid (PFOS) – C8F17SO2OH, and perfluorooctanoic acid (PFOA) – C7F15COOH. For this purpose, density functional theory (DFT) and its time-dependent formalism (TD-DFT) at both CAM-B3LYP/6-311+G(2d,2p) and M06-2X/6-311+G(2d,2p) levels of theory in water (IEF-PCM) were employed. To gauge the accuracy of the DFT approaches for the current systems, wave function methods (Møller–Plesset, MP2, coupled-cluster with single and double excitations, CCSD, CCSD with perturbative triples, CCSD(T), and equation of motion CCSD, EOM-CCSD) and aug-cc-pVXZ (X = D and T) basis sets were used. Regarding PFBS and PFOS, all the excited states probed were found to be energetically accessible only in the high-energy vacuum UV region (<200 nm ≥6.20 eV); SO2O− is released when the first low-lying excited singlet state (21A) of both compounds is accessed. On the other hand, two lowest-lying excited singlet states of PFOA were computed at considerably lower energy (5.84 eV and 5.97 eV for 21A and 31A, respectively, at the TD-DFT/CAM-B3LYP/6-311+G(2d,2p)). In addition, intramolecular OH radical formation is suggested for protonated PFOA when interacting with radiation at 7.98 eV ≈ 155 nm, as determined at the TD-DFT/CAM-B3LYP/6-311+G(2d,2p) level of theory. Such intramolecularly generated hydroxyl may contribute to a faster degradation of PFOA (or of other per- and poly-fluoroalkyl substances (PFAS) that are usually found together with PFOA).
 Please wait while we load your content...
Please wait while we load your content...

