
Solvation and the secondary structure of a proline-containing dipeptide: insights from VCD spectroscopy†

Abstract
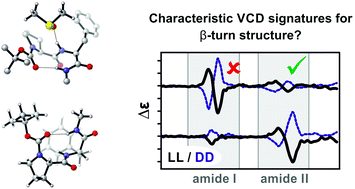
In this study we investigate the IR and VCD spectra of the diastereomeric dipeptide Boc-Pro-Phe-(n-propyl) 1 in chloroform-d1 (CDCl3) and the strongly hydrogen bonding solvent dimethylsulfoxide-d6 (DMSO-d6). From comparison of the experimental spectra, the amide II spectral region is identified as marker signature for the stereochemistry of the dipeptide: the homochiral LL-1 features a (+/−)-pattern in the amide II region of the VCD spectrum, while the amide II signature of the diastereomer LD-1 is inverted. Computational analysis of the IR and VCD spectra of LL-1 reveals that the experimentally observed amide II signature is characteristic for a βI-turn structure of the peptide. Likewise, the inverted pattern found for LD-1 arises from a βII-turn structure of the dipeptide. Following a micro-solvation approach, the experimental spectra recorded in DMSO-d6 are computationally well reproduced by considering only a single solvent molecule in a hydrogen bond with N–H groups. Considering a second solvent molecule, which would lead to a cleavage of intramolecular hydrogen bonds in 1, is found to give a significantly worse match with the experiment. Hence, the detailed computational analysis of the spectra of LL- and LD-1 recorded in DMSO-d6 confirms that the intramolecular hydrogen bonding pattern, that stabilizes the β-turns and other conformations of LL- and LD-1 in apolar solvents, remains intact. Our findings also show that it is essential to consider solvation explicitly in the analysis of the IR and VCD spectra of dipeptides in strongly hydrogen bonding solvents. As the solute–solvent interactions affect both conformational preferences and spectral signatures, it is also demonstrated that this inclusion of solvent molecules cannot be circumvented by applying fitting procedures to non-solvated structures.
 Please wait while we load your content...
Please wait while we load your content...

