
Probing radical–molecule interactions with a second generation energy decomposition analysis of DFT calculations using absolutely localized molecular orbitals†

Abstract
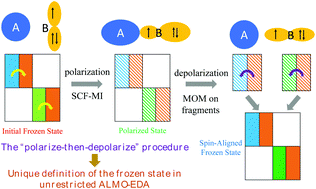
Intermolecular interactions between radicals and closed-shell molecules are ubiquitous in chemical processes, ranging from the benchtop to the atmosphere and extraterrestrial space. While energy decomposition analysis (EDA) schemes for closed-shell molecules can be generalized for studying radical–molecule interactions, they face challenges arising from the unique characteristics of the electronic structure of open-shell species. In this work, we introduce additional steps that are necessary for the proper treatment of radical–molecule interactions to our previously developed unrestricted Absolutely Localized Molecular Orbital (uALMO)-EDA based on density functional theory calculations. A “polarize-then-depolarize” (PtD) scheme is used to remove arbitrariness in the definition of the frozen wavefunction, rendering the ALMO-EDA results independent of the orientation of the unpaired electron obtained from isolated fragment calculations. The contribution of radical rehybridization to polarization energies is evaluated. It is also valuable to monitor the wavefunction stability of each intermediate state, as well as their associated spin density profiles, to ensure the EDA results correspond to a desired electronic state. These radical extensions are incorporated into the “vertical” and “adiabatic” variants of uALMO-EDA for studies of energy changes and property shifts upon complexation. The EDA is validated on two model complexes, H2O⋯˙F and FH⋯˙OH. It is then applied to several chemically interesting radical–molecule complexes, including the sandwiched and T-shaped benzene dimer radical cation, complexes of pyridine with benzene and naphthalene radical cations, binary and ternary complexes of the hydroxyl radical with water (˙OH(H2O) and ˙OH(H2O)2), and the pre-reactive complexes and transition states in the ˙OH + HCHO and ˙OH + CH3CHO reactions. These examples suggest that this second generation uALMO-EDA is a useful tool for furthering one's understanding of both energetic and property changes associated with radical–molecule interactions.
- This article is part of the themed collections: PCCP Perspectives and 2020 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

