
Modelling the enthalpy change and transition temperature dependence of the metal–insulator transition in pure and doped vanadium dioxide
 a
Yuzheng
Guo
*ac
and
a
Yuzheng
Guo
*ac
and
Abstract
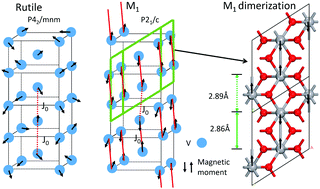
We compare various calculation methods to determine the electronic structures and energy differences of the phases of VO2. We show that density functional methods in the form of GGA+U are able to describe the enthalpy difference (latent heat) between the rutile and M1 phases of VO2, and the effect of doping on the transition temperature and on the band gap of the M1 phase. An enthalpy difference of ΔE0 = −44.2 meV per formula unit, similar to the experimental value, is obtained if the randomly oriented spins of the paramagnetic rutile phase are treated by a non-collinear spin density functional calculation. The predicted change in the transition temperature of VO2 for Ge, Si or Mg doping is calculated and is in good agreement with the experiment data.
- This article is part of the themed collection: 2020 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

