
Efficient sampling of high-energy states by machine learning force fields

Abstract
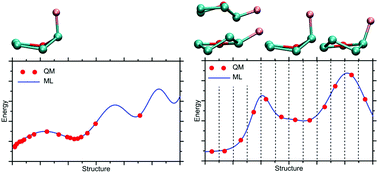
Regarding their application in the field of molecular sciences, machine learning (ML) methods are capable of combining the high accuracy of ab initio potentials with an efficiency closer to that of classical molecular mechanics. By relying on the reference data (e.g., atomic configurations and corresponding energies), the ML algorithms can reconstruct the potential energy surface for simple molecular systems, which may subsequently serve as a computationally inexpensive force field. The accuracy of such an ML force field is highly dependent on the character of the dataset that was used for its training. In this work, we show that omitting the high-energy states, which results from following the Boltzmann distribution, may lead to a catastrophic loss of accuracy in certain regions of the configurational phase space. To overcome this challenge, we have proposed an alternative solution for generating the ML input data. The most essential step is the biased subsampling of the configurations, aimed at increasing the population of hardly accessible states, usually located on energy barriers. The applicability of the proposed procedure is demonstrated on the example of conformational rearrangements in the two flexible, heterocyclic molecules. This approach provides an essential component required to obtain the ML force fields, accurate within the whole configurational phase space of the system.
 Please wait while we load your content...
Please wait while we load your content...

