
Solvation of coronene oligomers by para-H2 molecules: the effects of size and shape†
 *a
and
E.
Yurtsever
b
*a
and
E.
Yurtsever
b
Abstract
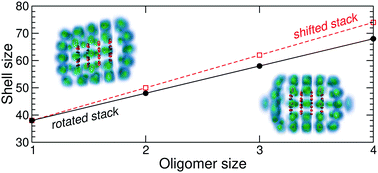
The stepwise solvation of various cationic coronene oligomers by para-hydrogen (p-H2) molecules was computationally investigated using a united-atom model for the p-H2 molecules and the Silvera–Goldman potential, together with a polarizable description for the interaction with the hydrocarbon molecules. A survey of the energy landscape for oligomers containing between 1 and 4 coronene molecules and possible different conformers was carried out using standard global optimization, the hydrocarbon complex being kept as rigid. The most stable structures provided the starting configuration of systematic path-integral molecular dynamics simulations at 2 K. The variations of the geometric and energetic properties of the solvation shell were determined with increasing number of para-hydrogen molecules. The relative stability of the solvation shell is generally found to be more robustly determined by the energy increment (or dissociation energy) than by geometrical indicators, especially when the oligomers have less ordered structures. In agreement with recent mass spectrometry experiments, the size at which the first solvation shell is complete is found to vary approximately linearly with the oligomer size when the coronene molecules stack together, with a slope that is related to the offset between two successive molecules.
- This article is part of the themed collection: 2020 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

