
First principle investigation of the influence of sulfur vacancies on thermoelectric properties of single layered MoS2
 *a
*a
Abstract
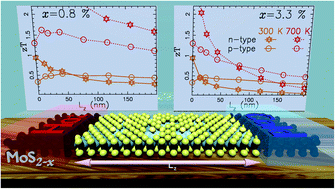
Thermoelectric properties of single layered transition metal dichalchogenide MoS2 are investigated on the basis of ab initio calculations combined with Landauer formalism. The focus is made on sulfur vacancy defects that are experimentally observed to be largely present in materials especially in exfoliated two dimensional MoS2 compounds. The impact of these defects on phonon and electron transport properties is investigated here using a realistic description of their natural disordering. It is observed that phonons tend to localize around defects which induce a drastic reduction in thermal conductivity. For p type doping the figure of merit is almost insensitive to the defects while for n type doping the figure of merit rapidly tends to be zero for the increasing length of the system. These features are linked with a larger scattering of the electrons in the conduction band than that of holes in the valence band.
 Please wait while we load your content...
Please wait while we load your content...

