
Prediction of optoelectronic properties of Cu2O using neural network potential†
 a
and
Pere
Miró
*a
a
and
Pere
Miró
*a
Abstract
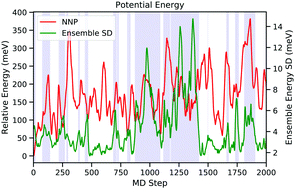
Neural network potentials (NNPs) trained against density functional theory (DFT) are capable of reproducing the potential energy surface at a fraction of the computational cost. However, most NNP implementations focus on energy and forces. In this work, we modified the NNP model introduced by Behler and Parrinello to predict Fermi energy, band edges, and partial density of states of Cu2O. Our NNP can reproduce the DFT potential energy surface and properties at a fraction of the computational cost. We used our NNP to perform molecular dynamics (MD) simulations and validated the predicted properties against DFT calculations. Our model achieved a root mean squared error of 16 meV for the energy prediction. Furthermore, we show that the standard deviation of the energies predicted by the ensemble of training snapshots can be used to estimate the uncertainty in the predictions. This allows us to switch from the NNP to DFT on-the-fly during the MD simulation to evaluate the forces when the uncertainty is high.
 Please wait while we load your content...
Please wait while we load your content...

