
Dissociative chemisorption of O2 on Agn and Agn−1Ir (n = 3–26) clusters: a first-principle study†
 *ab
*ab
Abstract
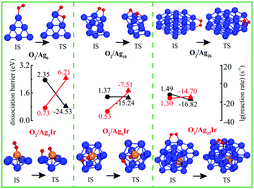
Understanding the interactions between O2 and small metal clusters is of great importance in exploring heterogeneous catalysis particularly involving an oxidation reaction. We herein present the dissociative chemisorption of O2 on Agn and Agn−1Ir clusters (n = 3–26) by using density functional theory calculations. Combining a particle swarm optimization algorithm and a minima hopping method, we have optimized and obtained stable geometric structures of Agn and Agn−1Ir clusters without and with O2 adsorption. Some important physical parameters, including bond length, adsorption energy, dissociation barriers and bader charge, have been systematically calculated for appraising the stability and reactivity of Agn and Agn−1Ir clusters. It is found that the dopant Ir atom can largely enhance the stability and promote the O2 dissociation, especially on small Agn−1Ir clusters (n = 3–10). It is mainly attributed to the dopant Ir atom being completely exposed outside the Ag atoms. For O2 adsorption and dissociation on large Agn−1Ir clusters (n = 11–26), the dissociation barriers are much higher due to the dopant Ir emerging into the core of Agn−1Ir clusters, which is very similar to those on large Agn (n = 11–26). Microkinetic simulation results provide direct evidence for high reaction temperature and pressure effects on improving O2 dissociation on Agn and Agn−1Ir clusters especially for small clusters (n < 10). It is found that the Ag5Ir cluster is the most suitable nanocluster for promoting O2 dissociation at the given reaction temperatures and pressures. Our theoretical work is helpful for the rational design of doped silver nanocluster catalysts in future experiments.
 Please wait while we load your content...
Please wait while we load your content...

