
Remarkable reversal of 13C-NMR assignment in d1, d2 compared to d8, d9 acetylacetonate complexes: analysis and explanation based on solid-state MAS NMR and computations†
 a
Ari
Pyykkönen,
b
Hans Jørgen Aa.
Jensen,
a
Vickie
McKee,
ac
Juha
Vaara
b
and
Ulla Gro
Nielsen
*a
a
Ari
Pyykkönen,
b
Hans Jørgen Aa.
Jensen,
a
Vickie
McKee,
ac
Juha
Vaara
b
and
Ulla Gro
Nielsen
*a
Abstract
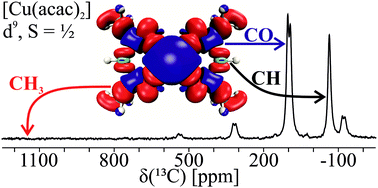
13C solid-state MAS NMR spectra of a series of paramagnetic metal acetylacetonate complexes; [VO(acac)2] (d1, S = ½), [V(acac)3] (d2, S = 1), [Ni(acac)2(H2O)2] (d8, S = 1), and [Cu(acac)2] (d9, S = ½), were assigned using modern NMR shielding calculations. This provided a reliable assignment of the chemical shifts and a qualitative insight into the hyperfine couplings. Our results show a reversal of the isotropic 13C shifts, δiso(13C), for CH3 and CO between the d1 and d2versus the d8 and d9 acetylacetonate complexes. The CH3 shifts change from about −150 ppm (d1,2) to roughly 1000 ppm (d8,9), whereas the CO shifts decrease from 800 ppm to about 150 ppm for d1,2 and d8,9, respectively. This was rationalized by comparison of total spin-density plots and computed contact couplings to those corresponding to singly occupied molecular orbitals (SOMOs). This revealed the interplay between spin delocalization of the SOMOs and spin polarization of the lower-energy MOs, influenced by both the molecular symmetry and the d-electron configuration. A large positive chemical shift results from spin delocalization and spin polarization acting in the same direction, whereas their cancellation corresponds to a small shift. The SOMO(s) for the d8 and d9 complexes are σ-like, implying spin-delocalization on the CH3 and CO groups of the acac ligand, cancelled only for CO by spin polarization. In contrast, the SOMOs of the d1 and d2 systems are π-like and a large CO-shift results from spin polarization, which accounts for the reversed assignment of δiso(13C) for CH3 and CO.
 Please wait while we load your content...
Please wait while we load your content...

